Gesetzgebung für Medizinprodukte in Deutschland
Startseite » Allgemeine Themen für Medizinprodukte » Medizinprodukte-Gesetzgebung in der EU und Deutschland
Einführung
In Deutschland gibt es für Medizinprodukte eine Vielzahl von Gesetzen, Verordnungen, einschlägigen Normen sowie Handlungsempfehlungen. Um einen Überblick zu gewinnen, ist es sinnvoll einen Blick auf die verschiedenen Akteure, Ihre Rolle und somit auf Ihre Absichten und Ziele zu erhalten.
Das Deutsche Medizinprodukte-Recht basiert auf der durch den Europäischen Rat sowie das Europäische Parlament verabschiedete MDR (Verordnung EU 2017/745 über Medizinprodukte), die unmittelbares Recht in den Mitgliedsländern der EU darstellt. Dies erfolgte wie in den Erwägungsgründen der MDR ersichtlich, mit dem Ziel der Harmonisierung des Medizinprodukterechts in Europa.


Verordnungen, Gesetze und Richtlinien - ein Überblick
EU-Recht
Bei der Umsetzung einer Richtlinie (vgl. abgelöste MDD) verfügt jedes Mitgliedsland über Freiräume in der Umsetzung. Hierdurch eine weniger homogene Medizinprodukte-Rechtslandschaft in Europa entstand. In Deutschland erfolgte die Umsetzung der MDD durch das Medizinproduktegesetz, das im Rahmen des Inkrafttretens der MDR zurückgezogen wurde.


EU-Verordnungen können durch Beschlüsse – sogenannte „modifications“ und „corrections“ – ergänzt und verändert werden. Diese können für die jeweilige Verordnung bzw. Richtlinie auf der Website des Amtes für Veröffentlichungen der EU aufgerufen werden.
Die Verordnungen werden durch Leitfäden ergänzt, die zwar nicht verbindlich sind, jedoch im Rahmen von Audits durch Behörden oder benannte Stellen faktische Wirkung entfalten. Die Wirkung beruht darauf, dass sie als anzuwendender Stand von Wissenschaft und Technik angesehen werden.
Globale Hamonisierung
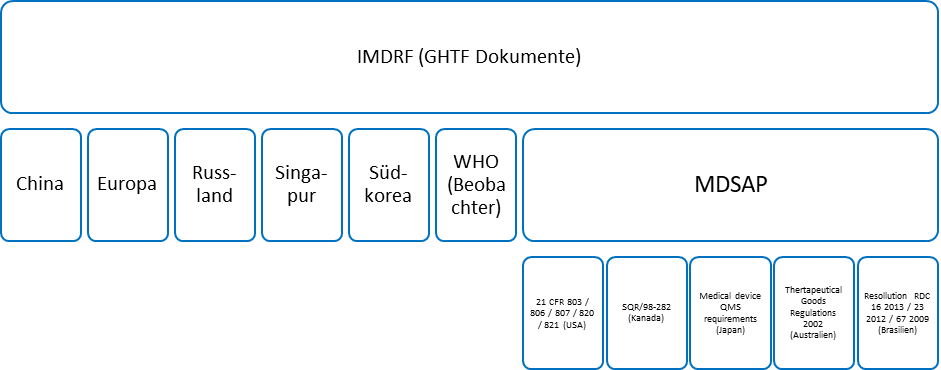
In Europa sind die Leitfäden der EU Kommission vorherrschend, welche in den MDCG Dokumenten (früher MEDDEV Dokumente – eine Vielzahl ist in Umstellung) niedergeschrieben sind. Diese wiederum fließen in die internationalen Leitfäden-Dokumente der IMDRF ein.
Zahlreiche Abschnitte der IMDRF und MDCG/MEDDEV Dokumente sind identisch. Dies ist Ausdruck der fortschreitenden globalen Harmonisierung.
Best practice Dokumente entfalten eine vergleichbare Wirkung, werden jedoch nicht von den Hauptorganen der EU, sondern Gremien wie der NBOG (Notified Body Operations Group) veröffentlicht. Diese richten sich explizit an die Benannten Stellen und bieten somit eine gute Quelle um das Vorgehen einer Benannten Stelle im Rahmen der Konformitätsbewertung sowie bei Audits zu verstehen. Unternehmen können in Vorbereitung auf die Zusammenarbeit mit der benannten Stelle mithilfe NBOG Dokumente die Ziele und Schwerpunkte der benannten Stelle verstehen und so zielgerichtet mit der benannten Stelle zusammenarbeiten.
Im Rahmen der IMDRF und auf Initiative der US amerikanischen FDA, haben sich 5 Behörden im MDSAP Programm zusammengeschlossen, um einen global gültigen Audit-Leitfaden zu erstellen. Das MDSAP Programm umfasst die Anforderungen der ISO 13485 sowie die nationalen Regelung der USA, Kanadas, Japans, Brasiliens und Australiens. Die Länder haben sich im Rahmen der nationalen Gesetz- und Verordnungsgebung zur gegenseitigen Anerkennung der Audits nach dem MDSAP Standard in einem definierten Rahmen verpflichtet. Das MDSAP Programm wird von den Behören der EU, des Vereinigten Königreichs sowie der WHO offiziell beobachtet und von sogenannten „affiliate members“ (Partner-Mitgliedern) in ihren Ländern anerkannt. MDSAP stellt somit den global umfassendsten und einheitlichsten Anforderungskatalog für das Qualitätsmanagement-System von Medizinprodukte-Herstellern dar.
Schließlich haben (harmonisierte) Normen Einfluss auf die umzusetzenden Vorgaben. Sie sind im Rahmen der Ermittlung des Stand der Technik im CE-Kennzeichnungsverfahren zu nutzen und bei Übereinstimmung mit den jeweils einschlägigen Normen wird die Konformität vermutet (Artikel 8 sowie Anhang 10 der MDR).
Deutsches Recht
Obgleich Verordnungen der EU unmittelbar in jedem Mitgliedsland wirksam werden, können sie durch nationale Gesetze ergänzt werden. Dies Ergänzung ist in Teilen notwendig um beispielsweise die in der MDR genannten „zuständigen Behörden der Mitgliedsländer“ zu benennen, reflektiert jedoch auch den Freiheitsgrad, den sich die Mitgliedsländer der EU erwarten. In Deutschland erfolgt die Ergänzung der MDR vorwiegend durch das Gesetz zur Anpassung des Medizinprodukterechts an die Verordnung (EU) 2017/745 und die Verordnung (EU) 2017/746 (Medizinprodukte-EU-Anpassungsgesetz – MPEUAnpG) sowie das Gesetz zur Durchführung unionsrechtlicher Vorschriften betreffend Medizinprodukte (MPDG).


* Gemäß § 85 MPEUAnpG: Zuständige Behörden im Sinne der Verordnung (EU) 2017/745 […] sind die Behörden der Länder – Beispiele:
- Baden-Württemberg: Regierungspräsidien (https://rp.baden-wuerttemberg.de/themen/gesundheit/medizinprodukte/)
- Hamburg: Behörde für Justiz und Verbraucherschutz https://www.hamburg.de/medizinprodukte/12279058/inverkehrbringen-sicherer-medizinprodukte/
- Berlin: Landesamt für Gesundheit und Soziales (LAGeSo) https://www.berlin.de/lageso/gesundheit/pharmaziewesen-und-medizinprodukte/medizinprodukte/
§ 85 MPEUAnpG beschreibt die zuständigen Behören im Sinne der MDR in Deutschland. Gemäß dem föderalen Prinzip sind dies die Behören der Länder. Somit bestimmt jedes Bundesland darüber, welche Behörde für Medizinprodukte zuständig ist. Während beispielhaft in Hamburg die Behörde für Justiz und Verbraucherschutz zuständig ist, sind es in Baden-Württemberg die 4 Regierungspräsidien mit ihren Gesundheitsämtern und in Berlin das LAGeSo.
Die ZLG
Dabei übernimmt die ZLG (Zentralstelle der Länder für Gesundheitsschutz bei Arzneimitteln und Medizinprodukten) einige Aufgaben der Länder, die im Staatsvertrag „Abkommen über die Zentralstelle der Länder für Gesundheitsschutz bei Arzneimitteln und Medizinprodukten“ geregelt sind. Dies sind insbesondere gemäß Artikel 2 Abs. 2 Nr. 1 die Benennung der benannten Stellen und gemäß Artikel 2 Abs. 4 Nr. 1 die Koordinierung der Weiterentwicklung des Qualitätssicherungssystems der Medizinprodukteüberwachung. Die ZLG dient auch als zentrale Ansprechstelle für alle Belange der EU, der Partnerländer sowie dem Kontakt mit internationalen Behörden. Die Akkreditierung der benannten Stellen obliegt gemäß § 1 Abs. 1 des Akkreditierungsstellengesetz (AkkStelleG) der Deutschen Akkreditierungsstelle GmbH (DAkkS). Die ZLG fungiert hier jedoch als Begutachter und Überwacher im Bereich Medizinprodukte im Auftrag der DAkkS. Die ZLG verfügt demnach über eine starke Position in Deutschland, wenngleich sie gegenüber dem Medizinproduktehersteller nur selten auftritt.
Unterstrichen wird der Einfluss der ZLG durch den Vorsitz des ZLG Präsidenten bei der NBOG der EU. Es ist daher ratsam die Stellungnahmen und Veröffentlichungen der ZLG aufmerksam zu verfolgen um zukünftige Entwicklungen im Bereich der Überwachung von Medizinprodukten und den benannten Stellen in Erfahrung zu bringen.


Neben den Ländern und der ZLG spielt das Bundesgesundheitsministerium (BGM) ein zentrale Rolle in der deutschen Regelsetzung. Es bietet einen Überblick über die EU-Regelungen, die deutschen Gesetze und Verordnungen sowie Antworten auf häufige Fragen beispielsweise zur Medizinprodukte-Betreiberverordnung. Im BGM wurden das MPEUAnpG sowie das MPDG entworfen und in die Gesetzgebung eingebracht.
Eine Behörde des BGM ist das BfArM (Bundesinstitut für Arzneimittel und Medizinprodukte), welches das Meldewesen für Medizinprodukte verantwortet. So verantwortet das BfArM das Deutsche Medizinprodukte-Informations- und Datenbanksystem (DMIDS) und koordiniert die deutschen Beiträge zur europäischen EUDAMED Datenbank.
Zudem ist das BfArM, sofern notwendig, für die Genehmigung klinischer Prüfungen von Medizinprodukten zuständig.
Fazit:
- Deutsche Gesetze ergänzen die EU Verordnungen, die von Leitfäden ergänzt werden …. Es ist SEHR KOMPLEX
- Lesen Sie die Leitfäden – für die Umsetzung des Qualitätsmanagement empfehlen wir MDSAP
- Bleiben Sie neugierig und gehen Sie Schritt für Schritt durch den Paragraphen Dschungel
Respekt, wenn Sie sich bis hierin durch den Beitrag gelesen haben – wir empfehlen an dieser Stelle einen Kaffee oder ein Glas Wein.
Wie hilfreich war dieser Beitrag?
Klicken Sie auf die Sterne, um zu bewerten!
Durchschnittliche Bewertung 5 / 5. Anzahl Bewertungen: 1
Bisher keine Bewertungen! Seien Sie der Erste, der diesen Beitrag bewertet.
Uwe Philippeit
Dozent & Berater für Qualitätsmanagement. Leidenschaft für rechtliche Aspekte des QM. Experte für Lieferantenmanagement und CAPA. Liebt Zusammenhänge & das Big-Picture.
Lehrbeauftragter an der DHBW.


Neugierig geworden?
Besuchen Sie unsere Seminare für Medizinprodukte. Dort können Sie in angenehmer Atmosphäre die Theorie und Praxis des Qualitätsmanagements erlernen. Alles anhand von leicht verständlichen Beispielen und mit Begeisterung vorgetragen.
